

Abstract
Background: Targeting leukemic stem cells without detrimental effects on hematopoietic stem cells is a major goal for improving cure rates for acute myeloid leukemia (AML). Strategies include targeting leukemia specific mutations or pathways where leukemic cells are more dependent, leading to so-called synthetic lethality. One potential target for the latter is PRMT5, an arginine methyltransferase that methylates arginine on histones and a large number of non-histone proteins including components of the spliceosome. PRMT5 is essential for the maintenance of normal hematopoietic stem cells, through p53-dependent and independent mechanisms.
Aim: To determine the relative importance of PRMT5 in normal and leukemic stem cells using genetic and pharmacologic approaches.
Hypothesis: Survival of leukemic stem cells will be more dependent on PRMT5 than normal hematopoietic stem cells.
Results: Using a conditional-null allele, we deleted Prmt5 in two mouse models of AML; AML1-ETO and MLL-ENL. Deletion of Prmt5 in AML1-ETO leukemia dramatically improved survival, with relapse only occurring in the setting of Prmt5-expressing cells. In contrast, deletion of Prmt5 in MLL-ENL leukemia had little effect.
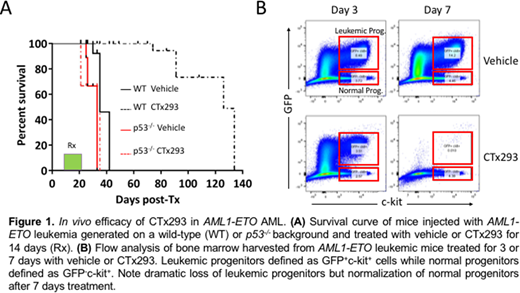
After screening a 350,000-compound library, we developed a potent and selective SAM-dependent inhibitor (CTx293) of PRMT5 similar to that reported by Chan-Penebre E. at al. Nat. Chem. Biol. 2015. We tested the in vivo activity of CTx293 in the AML1-ETO model generated on either a p53 wild-type or p53-/- background. Twice daily administration of CTx293 for 14 days demonstrated absolute p53 dependent activity, with prolongation of mean survival from 35 to 130 days (Figure 1A).
To directly compare the effects of PRMT5 inhibition on leukemic and normal progenitors, we examined the numbers of cells within the same animal during treatment with CTx293. After 3 days, there was a two-fold reduction in both leukemic and normal progenitors. However, after 7 days treatment, leukemic progenitors had reduced more than 1000-fold whilst normal progenitors (in the same mouse) had recovered (Figure 1B). To understand this differential effect on normal and leukemic progenitors, we FACS-isolated cells after 3 days therapy. While, there was evidence of p53 activation in both normal and leukemic progenitors, the downstream effects were quite distinct, with leukemic progenitors showing activation of apoptosis.
We tested the potency of CTx293 on primary human AML cells using a 14-day semi-solid agar growth assay. This demonstrated greater sensitivity of most AML samples (LD<30 nM) compared with healthy CD34+ cells (LD>100 nM). Of note, TP53-mutant samples were more resistant. Finally, we demonstrate activity of single agent CTx293 in 4 patient-derived xenografts.
Conclusion: We have used both genetic and pharmacologic approaches to show that PRMT5 is an attractive target for eliminating leukemic stem cells through a p53-dependent mechanism without toxicity to healthy stem cells.
Toulmin:CRC Cancer Therapeutics: Research Funding. Sonderegger:CRC Cancer Therapeutics: Research Funding. Cerruti:CRC Cancer Therapeutics: Research Funding. Street:CRC Cancer Therapeutics: Employment, Patents & Royalties; MERCK: Membership on an entity's Board of Directors or advisory committees. Stupple:CRC Cancer Therapeutics: Employment. Jane:CRC Cancer Therapeutics: Patents & Royalties. Wei:Novartis: Honoraria, Other: Advisory committee, Research Funding, Speakers Bureau; Celgene: Honoraria, Other: Advisory committee, Research Funding; Abbvie: Honoraria, Other: Advisory board, Research Funding, Speakers Bureau; Amgen: Honoraria, Other: Advisory committee, Research Funding; Pfizer: Honoraria, Other: Advisory committee; Servier: Consultancy, Honoraria, Other: Advisory committee, Research Funding. Altura:MERCK: Employment. Nicholson:MERCK: Employment. Curtis:MERCK: Membership on an entity's Board of Directors or advisory committees; CRC Cancer Therapeutics: Patents & Royalties, Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal